Open-sourcing of protein-structure software is already paying off
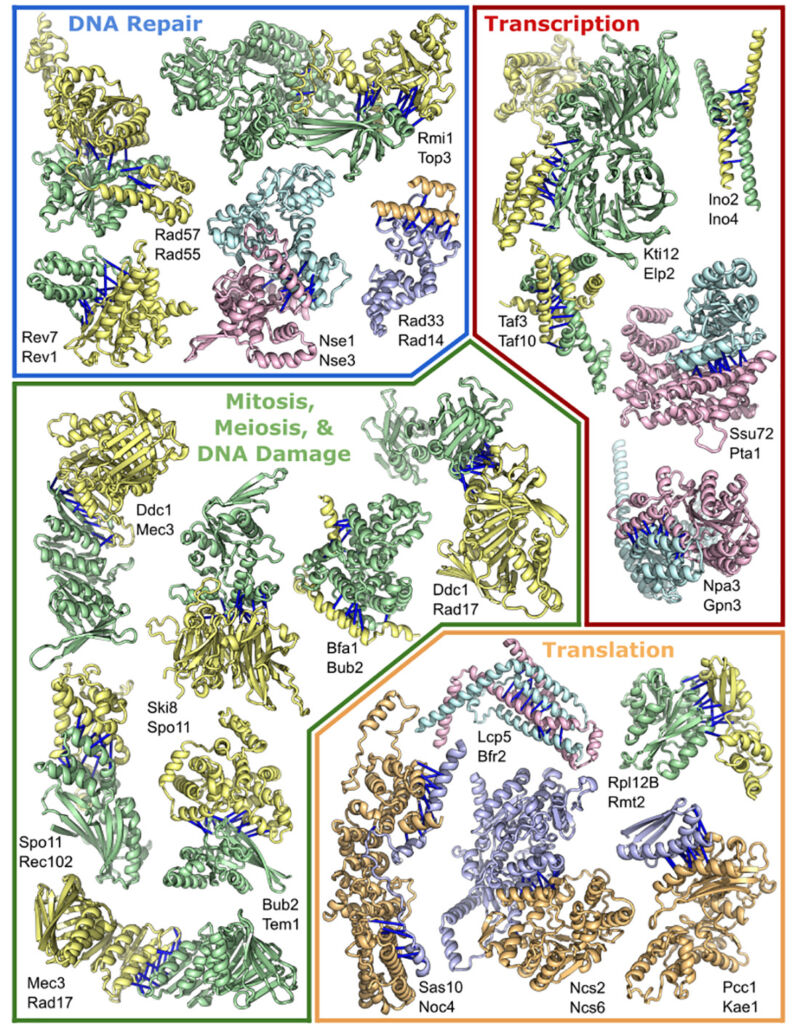
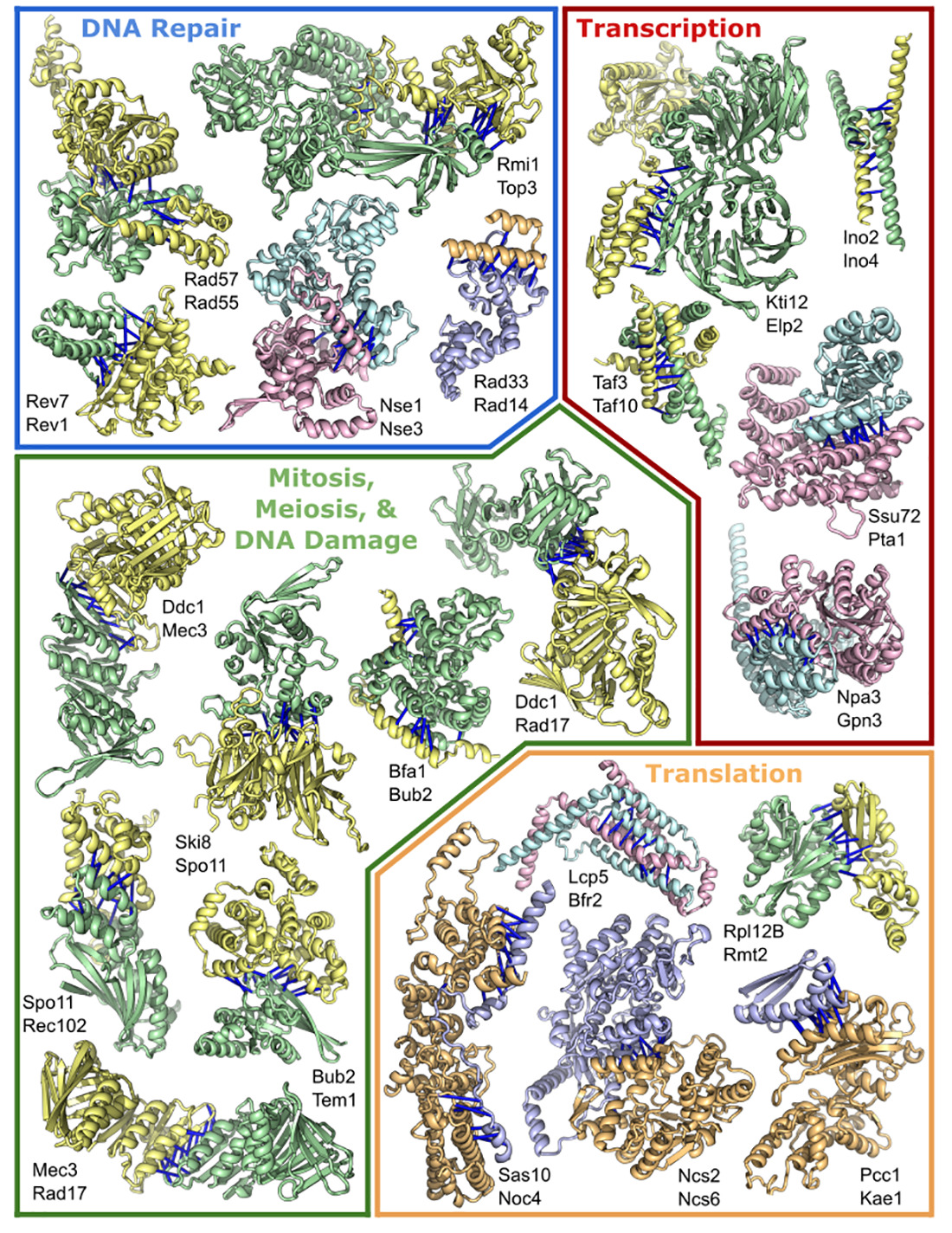
Enlarge (credit: Humphreys et. al.)
{kind=link}
It is now relatively trivial to determine the order of amino acids in a protein. Figuring out how that order translates to a complicated three-dimensional structure that performs a specific function, however, is extremely challenging. But after decades of slow progress, Google's DeepMind AI group announced that it has made tremendous strides toward solving the problem. In July, the system, called AlphaFold, was made open source. At the same time, a group of academic researchers released its own protein-folding software, called RoseTTAFold, built in part using ideas derived from DeepMind's work.
How effective are these tools? Even if they aren't as good as some of the statistics suggested, it's clear they're far better than anything we've ever had. So how will scientists use them?
This week, a large research collaboration set the software loose on a related problem: how these individual three-dimensional structures come together to form the large, multi-protein complexes that perform some of the most important functions in biology.
Read 19 remaining paragraphs | Comments